2 FILE STRUCTURE
To analyze real data using INTERQTL, some input data files are needed that include a pedigree file, a genome information file, files that contains marker genotypes for F1 parents and the progeny, respectively, and a file that contains trait records of the progeny. In simulation researches, however, only the pedigree file needs to be prepared manually. All other files are automatically generated using the simulation module.
The following is a brief guidance to preparing your input data files.
2.1 Input files
2.1.1 Genome file
This is a text file that contains information about chromosomes and markers. By default, the file name is given as “genome.txt”, but you may use whatever filename you like. Follow the following format to prepare this file.
[no_of_markers] (space) [no_of_chromosome] (space) [marker_name] (space) [map_position]
For example, a genome file for two chromosomes may look like as below. Be sure that a “-999” is put to end of the file. Otherwise, you’ll get warning of incorrect data format and the program will refuse to go on with the analysis. Also note that you must used underlines to connect words and digits for marker name.
0 0
0M__0 0
1 0 0M__1 10
2 0
0M__2 20
3 0
0M__3 30
4 0
0M__4 40
5 0
0M__5 50
6 0
0M__6 60
7 0
0M__7 70
8 0
0M__8 80
9 0
0M__9 90
10 0
0M_10 100
11 0
0M_11 110
12 0
0M_12 120
13 0
0M_13 130
14 0
0M_14 140
15 0
0M_15 150
16 1
1M__0 0
17 1
1M__1 10
18 1
1M__2 20
19 1
1M__3 30
20 1 1M__4
40
21 1
1M__5 50
22 1
1M__6 60
23 1
1M__7 70
24 1
1M__8 80
25 1
1M_19 90
26 1
1M_10 100
-999
2.1.2
Pedigree file
This is a text file that contains pedigree relationship in families that are used in the analysis. By default, the file name is given as “pedigree.txt”, but you may use whatever filename you like. An example of the pedigree file that contains 5 interconnected families goes below:
Type_of_inbred_cross DHL
Number_Of_mapping_families
5
Number_Of_founder_parents
5
Family_0_structure
20 0 1
Family_1_structure
20 1 2
Family_2_structure 20
2 3
Family_3_structure
20 3 4
Family_4_structure 20 4 0
The first line defines the type of progeny which is used in QTL mapping analysis. We use the following codes to define different progeny types:
BC1 ------ backcross to paternal
parent
BC2 ------ backcross to maternal
parent
F2 ------ intercross of F1s
DHL ------ double haploid lines
RIL ------ recombinant inbred lines
The second and third lines give the number of mapping families and the number of founder parents that these mapping families are derived. Note that we start counting families and parents from 0. Following lines are descriptions of each mapping family. In the right side, the numbers give family size (i.e. number of progeny in each family), paternal parent id and maternal parent id, respectively. For example, the first family (Family_0) consists of 40 progeny that are derived by mating parent_0 and parent_1. In preparing the file, you must also use underlines to connect words and digits for description phrases.
2.1.3 F1 marker file
This is a text file that contains marker genotypes for F1 parents. By default, the file name is given as “f1.txt”, but you may use whatever filename you like. Following the format below to prepare this file.
[marker_name] (space) [marker_0_genotype] (space) [marker_1_genotype] (space) ……
With the genome information given before, an example marker file for 5 f1 parents goes as below.
0M__0 01 12 23 34 40
0M__1 01 12
23 34 40
0M__2 01 12
23 34 40
0M__3 01 12
23 34 40
0M__4 01 12
23 34 40
0M__5 01 12
23 34 40
0M__6 01 12 23 34 40
0M__7 01 12
23 34 40
0M__8 01 12
23 34 40
0M__9 01 12
23 34 40
0M_10 01 12
23 34 40
0M_11 01 12
23 34 40
0M_12 01 12
23 34 40
0M_13 01
12 23 34 40
0M_14 01 12
23 34 40
0M_15 01 12
23 34 40
1M__0 01 12
23 34 40
1M__1 01 12
23 34 40
1M__2 01 12
23 34 40
1M__3 01 12
23 34 40
1M__4 01 12
23 34 40
1M__5 01 12
23 34 40
1M__6 01 12
23 34 40
1M__7 01 12
23 34 40
1M__8 01 12
23 34 40
1M__9 01 12
23 34 40
1M_10 01 12 23 34 40
Note that we code marker genotypes by their origins of founder parents. For example, genotype 01 means that the two marker alleles come from parent-1 and parent_1, respectively. Also, note that you have to use digits for coding marker genotypes. You cannot use characters for marker genotypes in INTERQTL.
2.1.4
Progeny marker file
This is a text file that contains marker genotypes for all progeny. By default, the file name is given as “markers.txt”, but you may use whatever filename you like. Following the example below to prepare this file.
{First
marker genotypes}
0M__1 00 11 11 11 00 11 00 00
00 11 00 11 11 11 11 00 00 00 11 00
{genotypes for individuals in family 0}
11 11 11 11 22 22 22 11 22 11 22
22 22 22 11 22 11 11 22 11 {genotypes
for individuals in family 1}
33 22 22 22 33 22 33 33 33 33 22 33 22 33 22
33 33 22 33 33 {genotypes for
individuals in family 2}
33 33 44 33 44 44 33 33 33 44
33 33 33 33 44 44 44 33 33 44
{genotypes for individuals in family 3}
00 00 00 44 00 00 44 44 00 44 00 00 00 44 00
00 44 00 00 00 {genotypes for
individuals in family 4}
{Second marker genotypes}
0M__2 00 11 11 11 00 11 00 00
00 11 00 11 11 11 11 00 00 00 11 00
{genotypes for individuals in family 0}
11 11 11 11 22 22 22 11 22 11 22 22 22 22 11
22 11 11 22 11 {genotypes for
individuals in family 1}
33 22 33 22 33 22 33 22 33 33 22
33 22 33 22 33 33 22 33 33 {genotypes
for individuals in family 2}
33 44 44 33 44 44 33 33 33 44 33 33 33 33 44
44 44 33 33 44 {genotypes for
individuals in family 3}
00 00 00 44 00 00 44 44 00 44
00 00 00 44 00 00 44 00 00 00
{genotypes for individuals in family 4}
{Third marker genotypes}
0M__3 00 00 11 11 00 11 00 00
00 11 00 11 11 11 11 11 00 00 11 00
{genotypes for individuals in family 0}
11 11 11 22 22 22 22 22 22
11 22 22 22 22 11 22 11 11 22 11
{genotypes for individuals in family 1}
33 22 22 33 33 22 33 22 33 33 22 33 22 33 22
22 33 22 33 33 {genotypes for
individuals in family 2}
33 44 44 33 44 44 33 33 33 44 33 33
33 33 44 44 44 33 33 44 {genotypes for
individuals in family 3}
00 00 00 44 44 00 00 44 00 44 00 00 00 44 00
00 44 00 00 00 {genotypes for
individuals in family 4}
……..
Provide data for marker genotypes locus by locus, with each line corresponding to each family. That is, the first line are marker genotypes for the first family, the second line for the second family, and so on. Note these above are genotypes for double haploids and heterozygous genotypes exist. ATTENTION: words in { } are comments and they should not be include in the trait file.
2.1.5
Quantitative trait file
This is a text file that contains quantitative trait values for all progeny. By default, the file name is given as “pheno.txt”, but you may use whatever filename you like. Following the example below to prepare this file.
{quantitative trait for individuals in the first family}
11.07
11.75 10.75 9.39
8.75 10.22 9.86
9.61 11.25 9.5
12.08 11 9.08
9.91 9.42 9.33
9.82 10.67 9.18
11.56
{quantitative trait for individuals in the second family}
9.16
7.88 10.27 10.63
10.06 10.36 10.23
11.73 10.8 10.67
11.32 10.82 10.66
11.09 9.48 11.26
8.34 10.24 9.24
9.69
{quantitative trait for individuals in the third family}
8.91 11.02 9.24
9.51 10.48 10.42
8.89 10.34 9.41
10.26
9.72 10.58 9.16
9.5 10.36 11.56
8.88 10.98 10.03
8.81
……
Provide quantitative trait values family by family, with the first 20 data as the trait values for the first family, the second 20 data as the trait values for the second family, and so on. Note that the order of individuals in each family should be in agreement in the marker genotype file and the quantitative trait file. ATTENTION: words in { } are comments and they should not be include in the trait file.
2.2 Output files
2.2.1
Files that contains location-wise posterior QTL
intensity or QTL variance
The analysis generates output files for location-wise posteriors of QTL intensity, and QTL variance if the QTL effect is random. By default, qtI100_xx.txt are files that contain location-wise posterior QTL intensity and qtlVar_xx.txt are files that location-wise posterior QTL variance, where xx is the number of replications in the analysis.
Following (Sillanpaa and Arjas,
1998), the evidences of QTL number and position were given in terms of
location-wise posterior QTL density. Briefly, we divided each chromosome into
intervals (bins) ![]() of equal length (say
2cM). The interval length reflects the resulting mapping resolution. Let
of equal length (say
2cM). The interval length reflects the resulting mapping resolution. Let
![]() (18)
(18)
be the approximate posterior QTL
intensity on interval ![]() obtained from the
Monte Carlo simulation, where S is the number of saved MCMC cycles
(sampling iterations), T is the number of putative QTL in the model, and
obtained from the
Monte Carlo simulation, where S is the number of saved MCMC cycles
(sampling iterations), T is the number of putative QTL in the model, and
![]() is the number of QTL
in
is the number of QTL
in ![]() in round t of
the simulation. The product
in round t of
the simulation. The product ![]() gives an
approximation of the posterior frequency of QTL in interval
gives an
approximation of the posterior frequency of QTL in interval ![]() .
.
For assessing QTL variance,
location-wise posterior densities for QTL variance are also defined. Let ![]() be the cumulative distribution
functions associated with QTL additive variance in the small interval
be the cumulative distribution
functions associated with QTL additive variance in the small interval ![]() , estimate of which is given as
, estimate of which is given as
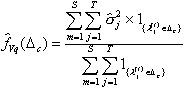 (19)
(19)
where ![]() is estimated variance
of QTL mapped to this interval.
is estimated variance
of QTL mapped to this interval.
The following are example files that contains location-wise posterior QTL intensity or QTL variance for 10 meta-runs. A QTL is simulated at 11 cM at the chromosome.
[Location-wise QTL intensity]
aType
1cM 3cM 5cM
7cM 9cM 11cM
13cM 15cM 17cM
19cM … …
1 0.62 0.87 1 2.42 18.91 27.63 5 0.85 0.79 1.19 … …
1 0.37 0.51 0.59
1.77 17.05 30.08
4.59 1.34 0.92 1.33 … …
1 0.34 0.51 0.63 1.62 20.24 27.23 4.87 0.95 0.69 1.02 … …
1 0.39 0.61 0.74 1.71
18.17 27.26 5.15 0.51 0.47 0.53 … …
1 0.49 1.08 1.57 2.18 19.72 27.05 5.02 1.02 0.99 1.24 … …
[Location-wise QTL variance]
aType
1cM 3cM 5cM 7cM 9cM 11cM 13cM 15cM 17cM
19cM … …
1 0.0617 0.0731 0.0745 0.1056
0.1399 0.1319 0.1178
0.1028 0.0931 0.1038
… …
1 0.0354 0.0413 0.0384 0.0850 0.1049 0.1062 0.1049 0.0949 0.0814 0.0711 … …
1 0.0692 0.0612 0.1048 0.1257
0.1567 0.156 0.1382 0.1055 0.0973 0.0979
… …
1 0.0681 0.0719 0.0757 0.1046
0.1651 0.1807 0.2244
0.1485 0.1189 0.0835
… …
1 0.0791 0.0983 0.1030 0.1234 0.1527 0.1771 0.1821 0.1453 0.0992 0.0819 … …
2.2.2 Files
that contain Markov chain values (i.e. posteriors of model parameters)
Users can also choose to generate files that contain Markov chain values (i.e. posteriors of model parameters). By default these files are named mix_xxMy where xx is the number of replications in the analysis and y is code for the type analysis.
The format of the mix files may vary, depending on settings of the analysis. Posterior parameters that may present in the mix files include:
logL --- the negative number is log likelihood of the model with an accepted QTL number.
QTL --- id of a putative QTL in the model
Chr --- id of chromosome where the putative QTL is on
qDist --- posterior location in cM of a putative QTL
nAl --- posterior number of alleles of a putative QTL
Sig(q) --- square root of the additive variance of the putative QTL
Var(e) --- residual variance
AV_Pxx --- additive value of the allele carried by parent xx
The following is an example mix file that contains 4 saved Markov chain values. Note that the positive numbers under the title of “logL” are number of iterations. The numbers that stand alone on the right are the numbers of currently accepted QTL in the model.
logL QTL
Chr qDist Sig(q)
Var(e) AV_P01 AV_P02
-418.84
1 1 12 0.5031 0.4549 0.1753
-0.1753 7
100600 2
1 41.92 0.9496 0 0.132 -0.132
100600 3
1 73.58 1.9748 0 -0.1209 0.1209
100600 4
1 102.94 3.7878 0 -0.2134 0.2134
100600 5
1 105.09 0.1034 0 0.1151 -0.1151
100600 6
1 122.02 3.2825 0 -0.1274 0.1274
100600 7
1 162.68 0.1179 0 -0.0722 0.0722
-435.15 1
1 11.52 1.2555 0.4965 0.1503 -0.1503 7
100700 2
1 39.53 1.3482 0 0.1468 -0.1468
100700 3
1 42.92 3.1662 0 -0.0114 0.0114
100700 4
1 78.86 3.4415
0 -0.1039 0.1039
100700 5
1 108.14 0.3116 0 -0.1014 0.1014
100700 6
1 127.15 3.8387 0 -0.1388 0.1388
100700 7
1 133.66 0.4084 0 -0.0176 0.0176
-428.87 1
1 10.92 1.1605
0.5461 0.1364 -0.1364 6
100800 2
1 43.32 1.6861 0 0.172 -0.172
100800 3
1 68.25 2.4239 0 -0.1233 0.1233
100800 4
1 76.92 3.8507 0
-0.0167 0.0167
100800 5
1 105.83 0.5681 0 -0.1263 0.1263
100800 6
1 125.96 3.7717 0 -0.1385 0.1385
-432.92 1
1 9.48 0.6776
0.4698 0.1342 -0.1342 7
100900 2
1 43.32 1.4168 0 0.1384 -0.1384
100900 3
1 69.21 2.0841 0 -0.1798 0.1798
100900 4
1 69.44 3.0075 0 0.0484 -0.0484
100900 5
1 94.6 0.0644 0 -0.0322 0.0322
100900 6
1 105.69 0.0632 0 -0.0554 0.0554
100900 7 1 125.4 3.5115 0 -0.1721 0.1721
… … … … … … … …